Python library for bioinformatics and scientific computing
这个包整合了我之前写过的一些用作分析和画图的脚本,做成 pip 包用起来会更方便,不需要输入路径去调用了,并且也可以作为基础来扩展更多的功能,现在有 agent 加持,写这种包还是很简单的。这里的脚本主要都是用于生信分析和可视化的,我起名字叫jsrc,用我的名字首字母开头,src就是源代码的意思了,jsrc 按功能分成模块,各模块之间没有依赖关系,想用哪个用哪个。
项目地址:https://github.com/imjiaoyuan/jsrc
使用 pip 安装:
pip install jsrc不过现在可能 uv 才是版本答案,可以自动解决依赖问题:
uv venv
uv run jsrc -h核心依赖只有 biopython 和 numpy,其他的按需安装:
pip install "jsrc[plot]" # 可视化(matplotlib + plotly)
pip install "jsrc[gs]" # 基因组选择(pandas + scikit-learn)
pip install "jsrc[vision]" # 图像分析(opencv-python)
pip install "jsrc[all]" # 全部都装CLI 入口是 jsrc,后面跟模块名和子命令。模块都使用懒加载,用到哪个才 import 哪个,启动速度不受模块数量影响。如果不想加载某些模块,可以通过环境变量控制:
export JSRC_DISABLE_MODULES=gs,vision # 禁用不用的模块
export JSRC_MODULES=seq,plot,analyze # 或者只启用某几个目前有 7 个模块,包括序列处理、可视化、基因组选择、基因调控网络展示、图像分析和后台任务管理:
seq 序列提取、翻译、QC、k-mer、滑窗分析
plot 基因/外显子/染色体/结构域/点图/环形图可视化
analyze 系统发育、motif、一致序列、SNP/INDEL、QC
gs 基因组选择数据集构建、划分、模型训练
grn 基因调控网络格式转换、中心性、交互式网络 viewer
vision 图像轮廓提取、椭圆傅里叶描述子、形态指标
job 后台任务提交、监控、日志、清理序列操作
这是最简单常用的模块,生信分析中反复出现的场景就是从基因组里提取特定区域啥的,seq 模块把这些操作统一成了几个子命令。
手里有基因组 FASTA、GFF 注释文件、以及一个基因 ID 列表,可以使用extra提取这些基因的指定类型序列,比如 CDS 序列:
jsrc seq extract -fa genome.fa -gff genes.gff -ids ids.txt -o cds.fa -feature CDS -match Parent这里 -feature 指定提取 CDS,也可以改成 gene、exon、mRNA 等,要看注释文件中对应的是什么。-match 是 GFF 属性里用来匹配 ID 列表的字段,也是要从注释文件中查看的。-match Parent 代表 ID 列表里的是基因名,而 GFF 的 CDS 行用 Parent 属性指向对应的基因。这个命令不是简单地找相同 ID 就提取,它会把同一个基因的多个 CDS 片段按照染色体坐标排序,合并重叠区域,再连成一条完整序列。遇到负链基因会自动取反向互补。输出是一个 FASTA,每个基因一条记录。
提取 CDS 后可以翻译成氨基酸序列:
jsrc seq translate -fa genome.fa -gff genes.gff -id ID -o proteins.fa它会解析 GFF 里的 CDS 特征,按基因分组、排序坐标、从基因组取序列,遇到负链自动反向互补,最后用 Biopython 翻译。碰到有移码的序列会跳过并给出警告。
不同来源的序列 ID 格式各不相同,命名方式五花八门,rename 可以把它们统一成自己想要的格式,支持 CSV 映射和 GFF 关联两种模式:
jsrc seq rename -fa in.fa -mode csv -map mapping.csv -o out.fa
jsrc seq rename -fa in.fa -mode gff -gff genes.gff -parent Parent -o out.faCSV 就是一个两列的映射表,第一列是原名,第二列是新的名称。GFF 模式则更适合从基因组注释做重命名,比如拿 mRNA 的 ID 去匹配它在 GFF 中的父基因名。
做顺式元件分析经常需要批量截取启动子。promoter 按基因列表一把全提出来,可以对每个基因指定上游和下游的长度:
jsrc seq promoter -fa genome.fa -gff genes.gff -ids genes.txt -o promoters.fa -up 2000 -down 0正链基因取起始密码子上游 N bp,负链基因取终止密码子下游 N bp(也就是上游)并反向互补。坐标超出染色体边界也会自动 clamp。
这是最轻量的 QC 工具,适合在进入复杂分析前快速看一眼序列的基本状况:
jsrc seq qc -fa assembly.faQC: 2 sequences, 268 bp total, GC 56.7%, N50 160.支持 FASTA(组装统计)和 FASTQ(测序统计),可以同时给:
jsrc seq qc -fa assembly.fa -fq r1.fq.gz r2.fq.gz -gs 520000000 --json如果给了 FASTQ 和基因组大小,还能估算测序深度。--json 可以输出结构化数据用于后续处理。FASTQ.GZ 也直接支持,不需要先解压。
从 CDS 序列计算密码子使用频率和 RSCU 值。RSCU > 1 表示该密码子使用偏多,< 1 表示偏少:
jsrc seq codon -fa cds.fa --top 20 --json它会按阅读框解析 CDS,跳过终止密码子和非 ACGT 字符,然后对每个氨基酸分别计算 RSCU。单条 CDS 或多条合并都可以。
k-mer 算是序列的"成分指纹",这个命令既可以对单条序列做高频 k-mer 统计,也可以在多个样本间算余弦距离:
# 单样本,看高频 k-mer
jsrc seq kmer -fa a.fa -k 7 --top 30
# 多样本,算两两余弦距离
jsrc seq kmer -fa a.fa b.fa c.fa -k 5多文件时输出的是一个距离矩阵,值在 0 到 1 之间,越接近 0 表示序列组成越相似。可以用来快速判断样本间是否大体一致。
GC 含量沿染色体的分布不是均匀的,滑窗分析能揭示局部变化。window 会在指定序列上做滑窗统计,输出每个窗口的 GC%、AT skew 和 GC skew:
jsrc seq window -fa genome.fa -id chr1 -w 1000 -s 200 --head 20GC skew 的正负可以指示复制起点和终止点的位置(前导链倾向于 G 多于 C),这在原核基因组分析中比较有用。
一些简单的分析操作
序列比对做完之后,常见操作就是建树、找 motif、看保守性、找差异。analyze 把这些串了起来,但是本着使用尽量少的依赖的原则(极简主义者癖好),所以目前实现的分析其实比较有限。
对一组比对好的或未比对的序列,用邻接法或 UPGMA 建一棵树,输出 Newick 格式。底层用 Biopython 的距离计算和建树模块:
# 邻接法(默认)
jsrc analyze phylo -fa aligned.fa -o tree.nwk
# UPGMA
jsrc analyze phylo -fa aligned.fa -a upgma -o tree.nwk输入序列未比对的话会自动做等长填充(短的补 -)。输出的 Newick 可以用任意树可视化软件查看。
phylo 的进阶版——bootstrap 重复采样给分支打上置信度。重复次数和随机种子都可以设置,保证结果可重复:
jsrc analyze bootstrap_phylo -fa seqs.fa -n 200 -seed 42 -o boot.nwk内部实现是先构建基准树,然后对多序列比对的列做有放回重采样,每次重采样重新建树,统计每个分支出现的频率作为 bootstrap 值。需要至少 3 条序列。
最直接的 motif 寻找方式——从序列中枚举所有 k-mer,按出现频率排序。虽然不如 MEME 这类专业工具精细,但胜在速度快、结果直观,适合先粗筛再决定要不要上更重的工具:
jsrc analyze motif -fa promoters.fa -o motif_out -nmotifs 5 -minw 6 -maxw 12从多序列比对中计算一致序列和每个位点的保守性分数。保守性 = 该位点出现频率最高的碱基的比例,数值越高表示该位点越保守:
jsrc analyze msa_consensus -fa aligned.fa --json可以用来快速判断比对质量:整体保守性高说明序列之间相似性好,适合做进化分析;如果整体保守性偏低,可能需要换个比对策略。
只比对两条序列,给出 SNP 和 INDEL 的详细统计。用 Biopython 的 PairwiseAligner 做全局比对:
jsrc analyze snpindel -fa pair.fa --json输出比对得分、对齐长度、匹配碱基数、SNP 数、INDEL 事件数和涉及的碱基数。非常适合做同一基因在不同样本间的快速比较。
这个命令可以同时接收 FASTA(组装统计)、SAM(比对统计)、VCF(变异统计)、FASTQ(测序统计),一次性给出多角度的质量概览:
jsrc analyze qc -fa assembly.fa -sam aln.sam -vcf variants.vcf.gz -fq r1.fq.gz r2.fq.gz -gs 520000000比 seq qc 多了解析 SAM 比对 CIGAR 计算深度和 VCF 区分 SNP/INDEL 的功能。适合在做完整套分析后做一次统一的质控汇总。
简单的可视化
做生信分析少不了要画图,不管是汇报还是文章。plot 模块把几种最常见的生物学图形封装成了命令行,输入 GFF 或 TSV 就可以出图,省去写 matplotlib 代码的时间。
所有图都输出 PNG,内核统一用 matplotlib Agg 后端,不需要显示器环境。
GFF + ID 列表画基因结构示意图。每个基因用一条水平线表示全长,蓝色方块表示 CDS 区域:
jsrc plot gene -gff genes.gff -ids ids.txt -o gene.png -dpi 300基因会按自然序(chr9 排在 chr10 前面,而不是字典序)纵向排列,适合展示多个基因的结构差异。
和 gene 类似,但突出外显子区域,用绿色区分:
jsrc plot exon -gff genes.gff -ids ids.txt -o exon.png -dpi 300如果你更关注外显子的数量和分布差异而不是整个 CDS,用 exon。
把基因在染色体上的位置可视化出来。读取 GFF 里的 ##sequence-region 元数据作为染色体长度,然后在对应位置画红色标记:
jsrc plot chromosome -gff genes.gff -ids ids.txt -o chr.png不加 -ids 时把所有基因都画上,加了就只标出目标基因。适合看基因在染色体上的聚集情况。
TSV 格式输入,每行是一个结构域,包含蛋白名、结构域名、起始和终止位置:
jsrc plot domain -tsv domains.tsv -o domain.png每个蛋白一条水平线,结构域用橙色方块标出并标注名称。适合快速查看结构域顺序和相对位置。
BED 格式输入,在染色体上标出顺式元件的分布:
jsrc plot cis -bed motifs.bed -o cis.png每条染色体画一条水平线,元件用蓝色竖线标出,并带名称标签。
用精确 k-mer 匹配画两条序列的点图。对角线上的点表示两序列的共线性区域,散落在外的点可能代表重复序列或重排:
jsrc plot dotplot -fa1 genome1.fa -fa2 genome2.fa -k 10 -o dotplot.png-k 控制 k-mer 大小:k 越小点越密但假阳性越多,k 越大特异性越高。不指定 -o 的话会弹出交互式窗口。
一个轻量级的 Circos 替代方案——输入 FASTA,画出环形染色体分布图,内圈显示 GC 含量滑窗统计:
jsrc plot circoslite -fa genome.fa -w 100000 -o circos.png-w 控制滑窗大小,默认 100 kb。外圈灰色环是染色体骨架,内圈蓝色柱子表示该窗口的 GC 比例。
还有两个是当作小彩蛋加的。heart 画一个动态心形曲线(matplotlib 动画),rose 画一个 3D 玫瑰曲面(Plotly 交互):
jsrc plot heart
jsrc plot rose没什么实用价值,纯属写着玩的。
基因组选择
基因组选择的分析流程相对固定:数据准备、划分交叉验证、模型训练和评估。gs 模块把这三个步骤做成了三个子命令,串成一条流水线。
输入 PLINK 二进制文件和表型文件,构建带模拟样本的数据集:
jsrc gs build -pheno phenotype.txt -plink /path/to/hap1 -o data/hap1 --n-sim 500 --top-k 2000 --h2 0.5 --seed 42内部流程是这样的:
- 先用 PLINK 做 LD pruning(
--indep-pairwise 50 5 0.2),得到近似独立的 SNP 集合 - 用
pandas-plink读取修剪后的基因型数据 - 对真实数据做 ANOVA F 检验筛选 top K 标记作为候选因果位点。
- 从真实样本中随机抽取两条作为"亲本",对每个标记随机选择来自亲本 1 或亲本 2,模拟子代基因型
- 用因果位点的线性组合计算育种值,添加高斯噪声控制遗传力(
--h2)。 - 输出
X.npy(基因型矩阵)、y.npy(表型)、sample_ids.txt和snp_ids.txt
模拟样本的遗传力参数很重要,h2=0.5 表示表型变异中一半由遗传因素解释,这在模拟中可以灵活调整来评估不同遗传力下的预测准确性。
把真实样本随机分成 K 折,模拟样本全部加入训练集:
jsrc gs split -i data/hap1 --folds 5 --seed 2024输出 cv_indices 目录,每折包含 fold_{i}_train.txt 和 fold_{i}_test.txt,分别记录行号。
在交叉验证上运行多个模型并比较结果:
jsrc gs train -i data/hap1 -o data/hap1/results --folds 5 --select-k 1000 --models gbdt,rf,et,lr,svm,nb支持 6 种 Python 模型:
| 代码 | 模型 | 主要参数 |
|---|---|---|
| gbdt | Gradient Boosting | 200 棵树,lr=0.05, max_depth=4 |
| rf | Random Forest | 300 棵树,class_weight=balanced |
| et | Extra Trees | 300 棵树,class_weight=balanced |
| lr | Logistic Regression | LBFGS, max_iter=2000 |
| svm | SVC | RBF 核,probability=True |
| nb | Gaussian Naive Bayes | — |
每个模型在每折上记录 accuracy、F1、MSE、AUC。最终输出 results.csv(逐折逐模型)和 summary.csv(各模型的均值和标准差),可以直接看哪个模型在这个数据集上表现最好。
基因调控网络可视化
这个是我自认为做的很好看的一个 grn 网络可视化。基因调控网络分析通常包含几个步骤,数据格式转换、计算网络统计量、可视化。grn 模块覆盖了这些环节,而且自带了完整的交互式网络浏览器。
GRN 的数据通常有散列:source、target、weight。net2json 把它们转成 JSON,启动 http 服务器以后可以动态加载,并且可以打包成独立的 ZIP 文件:
jsrc grn net2json -i network.tsv -o json/grn.json -n annotation.tsv -z viewer.zip -s展示模式有两种:
-a(all 模式):当网络中的基因数量小于等于阈值时,自动全量展示整张网络。适合几百个基因的小网络。-s(some 模式):初始只显示一个空画布,通过搜索基因名来展开其邻居。适合几千个基因的大网络,不会把浏览器卡死。
可选的 -n 参数接受一个注释 TSV,包含基因 ID、Potri ID 和功能描述,这些信息会在 viewer 里显示在节点的详情面板中(最开始给杨树做的所以有 Potri ID,后续会完善)。
想知道哪些基因在调控网络中处于核心位置,最直接的方式就是算度(degree)。这个命令统计每个节点的入度、出度和总度,按总度排序输出:
jsrc grn centrality -i network.tsv --top 30输出:
rank node in_degree out_degree total_degree
1 GENE_0504 24.14 34.97 59.12
2 GENE_0785 26.37 29.51 55.88
...入度高说明该基因被很多其他基因调控("听命令的"),出度高说明该基因调控很多其他基因("发号施令的")。总度排名靠前的往往是转录因子或信号通路的关键节点。
网络数据准备好之后,直接在本地起一个 HTTP 服务查看:
jsrc grn serve -d viewer -g json/grn.json -n json/annotation.json -p 8000 -s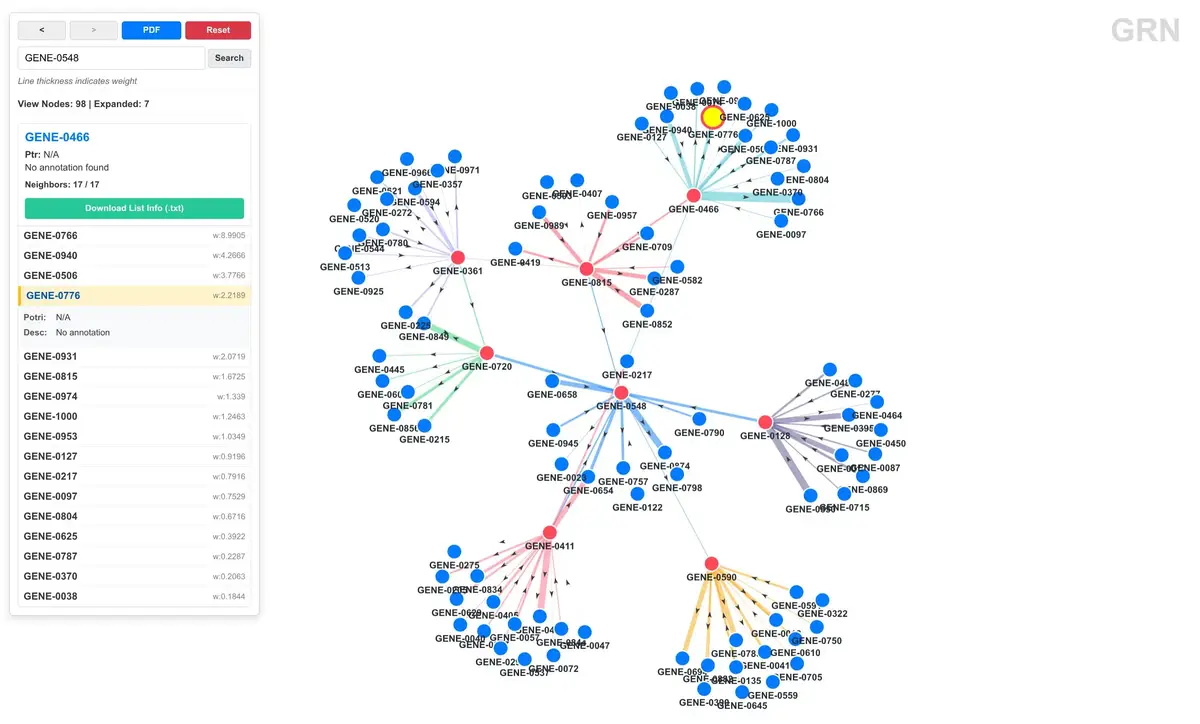
这个 viewer 是基于 D3.js 的 Force Graph 实现的交互式页面,功能包括:
- 力导向布局,节点自动排斥、边自动吸引
- 点击节点展开其 top 100 邻居(有边权排序)
- 搜索框可以快速定位到指定基因
- 信息面板显示基因的 Potri ID 和功能描述
- 邻居列表按边权降序排列,支持下载为 TXT
- PDF 导出(通过 html2canvas + jsPDF)
- 前进/后退导航历史
如果你只需要把注释 TSV 转成 JSON,不涉及网络转换,可以直接用这个:
jsrc grn anno2json -i annotation.tsv -o json/annotation.json图像分割与数值化
vision 模块主要面向植物叶片之类复杂图像的轮廓提取和形状分析,用 OpenCV 做底层的图像处理。
这是图像分析的第一步——从原始照片中提取出目标物体的轮廓。输入一张照片,输出每个物体的轮廓点集和边缘图:
jsrc vision extract -i leaf.jpg -o extracted/ --channel a --invert --save-mask处理流程:
- 高斯模糊降噪(
--blur控制核大小) - 提取指定通道(灰度、LAB a/b 通道、HSV s/v)
- Otsu 自动阈值二值化
- 形态学开运算去噪点 → 闭运算填孔洞
- 找外轮廓
- 按面积和长宽比筛选(排除太小的噪点和太大的背景)
- 按 x 或 y 坐标排序输出
通道选择对结果影响很大。绿色叶片在 LAB 色彩空间的 a 通道(绿-红轴)上对比度最好,比直接用灰度图鲁棒得多。输出两个文件:{name}_{i}_edge.png(轮廓)和 {name}_{i}.npy(轮廓点坐标数组)。
提取出轮廓之后,使用傅里叶级数逼近闭合轮廓,级数的系数可以作为形状的定量特征:
jsrc vision efd -i extracted/ -o descriptors/ --harmonics 20 --points 300EFD 的数学想法很简单:把轮廓的 x 坐标和 y 坐标分别看作弧长的周期函数,对它们做傅里叶展开。前几个谐波描述粗略形状,后面的谐波增加细节。通常 10-20 个谐波就能很好地重建大部分生物形状。
这个命令的输出是 {name}_efd.csv,包含 an、bn、cn、dn 四个系数的列。系数的数量 = 谐波数 × 4(比如 20 个谐波就有 80 个系数)。默认会生成一个对比图,把原始轮廓(黑色)和 EFD 重建轮廓(红色虚线)叠在一起展示。


系数做了归一化处理:旋转不变性(把第一谐波椭圆的长轴对齐到 0°)、缩放不变性、起始点归一化。这样即使照片里叶片的摆放角度、大小不同,同一种叶形的系数也是相近的。
做图像测量时经常需要计算面积、周长、圆度等指标。traits 命令从图像中提取最大物体的形态参数:
jsrc vision traits -i leaf.jpg --channel a --invertarea 461833.5
perimeter 3479.84
aspect_ratio 0.671
circularity 0.479
extent 0.535
solidity 0.837各指标的含义:
| 指标 | 含义 | 用途 |
|---|---|---|
| area | 像素面积 | 绝对大小 |
| perimeter | 轮廓周长 | 边界复杂度 |
| aspect_ratio | 宽高比 = 宽/高 | 细长程度 |
| circularity | 圆度 = 4π·area/perim² | 接近圆形的程度,1=正圆 |
| extent | 填充度 = area/外接矩形面积 | 形状的紧凑性 |
| solidity | 实心度 = area/凸包面积 | 边缘的锯齿/裂片程度 |
后台任务管理
经常跑任务的会遇到一个问题,如果用集群的 slurm 作业系统还好,但是小服务器上面使用 nohup 把任务挂后台的话,可能会分辨不出任务究竟是哪个、任务有没有正常完成、任务运行多长时间之类的问题,所以我做了这个job模块,做的事情就是把"nohup + 记录 PID + 定时查日志 + 收尾清理"这个流程标准化。
jsrc job submit "Rscript 02.harmony2.R" -N harmony -C /data/proj -S bash它会用 nohup 启动命令,同时记录 PID、启动时间、工作目录。每条记录自动分配一个 job_id。如果没指定日志路径,会自动在 ~/.local/share/jsrc/job-logs/{id}.log 创建一个。
这里在后台包装了一层——命令外面包了一个 echo $? > state_file 的尾随命令,用来捕获退出码。这样即使命令跑完了,也能区分正常退出(exit code 0)、失败(非 0)还是被杀死。
jsrc job ls -w -s rss_mb -r -l 20
# 只看特定任务
jsrc job ls -q harmony-w 模式会每 2 秒刷新一次,持续监控运行状态。列可以自定义选择,排序字段也很多——运行时长、RSS 内存(当前/平均/峰值)都可以当排序依据。
RSS 内存是从 /proc/{pid}/status 读的(Linux),对于追踪内存泄漏或确认任务是否正常运行很有用。ls 会记录每个任务的 RSS 最小值、峰值和平均值,而不是只存当前快照。
jsrc job show 12 -f json比 ls 更详细地展示单条记录的全部字段,适合精确定位问题。
# 看最后 200 行
jsrc job logs 1 -n 200
# 实时跟随输出
jsrc job logs 1 -F支持 follow 模式,类似 tail -f。方便在跑长任务时实时看输出。
# 温柔终止
jsrc job kill 12 -s TERM
# 暴力终止,连子进程一起杀
jsrc job kill 12 -s KILL -g-g 会把整个进程组杀掉,避免僵尸子进程残留。
jsrc job history -l 100 -f tsv默认保留最近 50 条,可以用 -l 扩展。除了不占用终端历史,好处是即使跨了终端会话、甚至机器重启了,记录还在。
时间久了积累的记录和状态文件越来越多,做一次清理:
jsrc job gc -k 1000 --prune-missing-log --remove-dead-state-k 是保留最近多少条历史,--prune-missing-log 标记日志文件已被删除的记录,--remove-dead-state 清理不在历史中的陈旧状态文件。
所有 job 数据存在 ~/.local/share/jsrc/jobs(也可以设 JSRC_JOBS_FILE 环境变量改路径),是一个 TSV 文件。每条记录包含:job_id、名称、提交时间、开始结束时间、状态、PID、退出码、工作目录、日志路径、RSS 统计、运行时长、原始命令。
状态机:running → exited(码 0)/ failed(非 0)/ killed(信号)/ lost(失踪)。
不足
最大的问题就是测试不全面,很多问题没有暴露出来,所以希望有更多人来用,我们一起找问题解决问题,如果有朋友有好的建议也可以找我,尽量方便更多人使用。
Talk about this post via [email protected]